 探討高濃電解液陰離子對Li+活度和嵌入反應動力學影響機理
探討高濃電解液陰離子對Li+活度和嵌入反應動力學影響機理
研究背景
鋰離子電池(LIBs)的電化學反應發生在電極/電解液界面,這些反應的動力學顯著影響LIBs的倍率性能。鋰離子在界面處發生插層和脫出,包括溶劑化和脫溶劑化。Ogumi和Abe等人的研究結果表明,Li+的脫溶是電荷轉移反應在電極/電解液界面上的決速步驟(RDS)。
界面電荷轉移反應的活化能與Li+離子和溶劑之間的相互作用相關,因此, Li+與溶劑間弱的相互作用有利于減少界面處的活化能,提高界面反應的速率。此外,美國陸軍實驗室的許康等人研究表明,由電解液的還原分解產物形成的石墨負極表面的固體電解液界面(SEI)也影響了石墨/電解液界面上Li+的插層動力學。因此,電解液組成對LIBs中的界面動力學至關重要。
近年來,含有超過3mol/L鋰鹽的高濃電解液(HCE)因其獨特的物理化學和電化學性質而逐漸受到研究人員的青睞。與1mol/L鋰鹽的傳統電解液相比, HCE中的Li+插層反應速率更快。通常,在Li鹽濃度高于3mol/L的HCE中,由于溶劑變少,Li+離子會與更多的陰離子配位以滿足其配位數。作者以前報道過HCE的Li+配位結構和物理化學特性受到陰離子物種的顯著影響。然而,陰離子種類對電極/電解液界面上電荷轉移動力學影響的機理仍不明朗。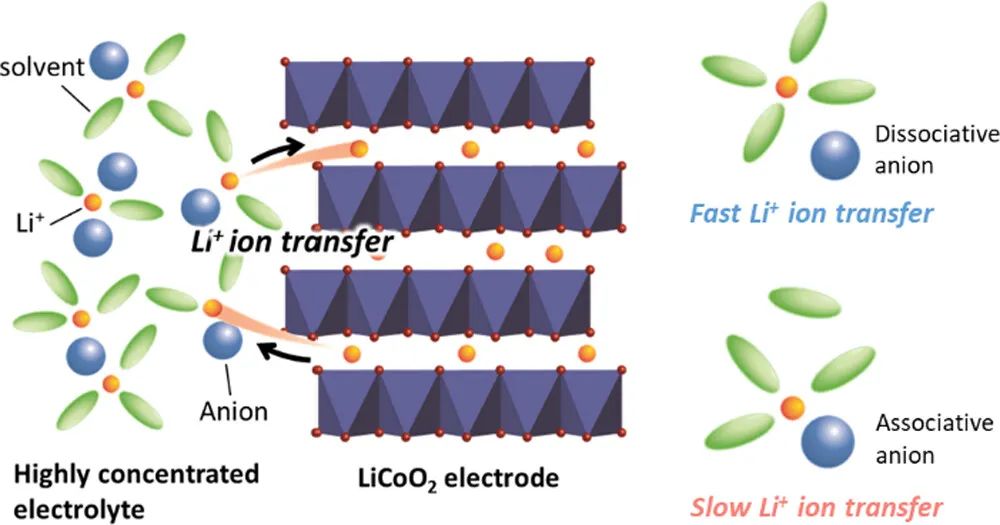
內容圖(TOC)
成功簡介
因此,Kaoru Dokko等人研究了陰離子對以碳酸丙烯酯(PC)為溶劑的高濃電解液中Li+溶劑化結構和電化學性能的影響。首先選取了5種常見的鋰鹽:六氟磷酸鋰(LiPF6)、雙(氟磺酰基)酰胺鋰(LiFSA)、雙(三氟甲磺酰基)胺鋰(LiTFSA)、高氯酸鋰(LiClO4)、四氟硼酸鋰(LiBF4)和三氟甲磺酸鋰(LiOTf)(圖1)。
在這項研究中,作者使用拉曼光譜分析了HCE的液體結構。使用concentration cell評估了HCEs中Li+的活度。最后,作者使用EIS研究了LiCoO2電極的Li+嵌入/脫嵌動力學。該工作以“Anionic Effects on Li-Ion Activity and Li-Ion Intercalation Reaction Kinetics in Highly Concentrated Li Salt/Propylene Carbonate Solutions”為題發表在The Journal of Physical Chemistry C上
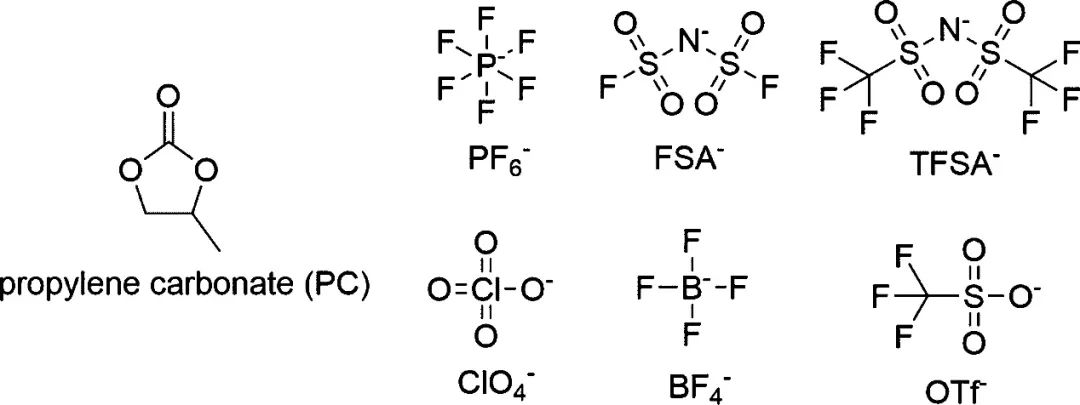
圖1. PC和Li鹽的陰離子的化學結構。
研究亮點
隨著HCEs中陰離子的Lewis堿性降低,Li+與陰離子之間的離子締合減弱,游離PC的濃度降低,導致Li+的活度系數更高。
在HCEs中使用具有弱Lewis堿性的陰離子的Li鹽有利于在LiCoO2/電解液界面上實現快速的電荷轉移動力學。
LiCoO2電極的穩定性可以使用具有弱Lewis堿性的陰離子HCE來改善。
圖文導讀
Li+在高濃PC電解液中的溶劑化結構
用拉曼光譜研究了不同濃度下PC電解液的液體結構。圖2顯示了680–780 cm–1范圍內 純PC和鋰鹽/PC電解液的拉曼光譜。純PC在712 cm–1處出現峰值,這是對稱環變形模式產生。當將Li鹽添加到PC中時,在~722 cm–1處出現一個新的峰,對應于與Li+離子配位的PC分子。游離和配位PC的拉曼帶強度提供了關于電解液的液體結構的信息。
圖2. 拉曼光譜顯示了在30°C下含有(a)LiPF6、(b)LiFSA、(c)LiTFSA、(d)LiClO4、(e)LiBF4和(f)LiOTf鹽的PC的對稱環變形模式。
圖3顯示了對應于陰離子振動帶的Li鹽/PC電解液的拉曼光譜。每個陰離子振動帶對Li+和電解液中相應陰離子之間的相互作用敏感。對于[LiClO4]/[PC]≤1/6的混合物,在~934 cm–1處觀察到ClO4–陰離子的對稱拉伸模式的峰,這是溶劑分離離子對(SSIP)中游離ClO4–的特征;這意味著[LiClO4]/[PC]≤1/6的混合物中的Li+離子被PC分子溶劑化。
圖3.(a)LiPF6(PF6對稱拉伸)、(b)LiFSA(S═O拉伸),(c)LiTFSA(CF3彎曲與S–N拉伸耦合)、(d)LiClO4(ClO4對稱拉伸)、(e)LiBF4(BF4對稱拉伸)和(f)LiOTf(SO3對稱拉伸)。在~722、778、960和1227 cm–1處由*表示的峰值分別對應于PC的對稱環變形、環變形、碳酸鹽對稱拉伸振動和碳酸鹽不對稱拉伸振動模式。
鋰金屬的電極電勢
為了評估電解液溶液中Li+離子的活度,測量了 Li 金屬的電極電勢。在鋰金屬的沉積/溶解過程中,Li+的溶劑化和去溶劑化發生在電極/電解液界面。因此,鋰金屬在非水電解液溶液中的電化學反應可描述如下:
電極電勢用能斯特方程表示如下
圖4. 30°C時,Li/Li+電極電位與PC電解液中Li鹽濃度的關系圖。參比電極為1 mol/L LiTFSA/PC中的Li/Li+。
即使Li沉積/溶解反應發生在Li金屬/SEI界面,鋰金屬的電極電勢也由本體電解液溶液的組成決定。圖4顯示了使用濃度池測量的各種鋰鹽/PC電解液中Li金屬的電極電勢。使用Concentration cell [Li|1M LiTFSA/PC||x M Li鹽/PC|Li]測試了樣品溶液中Li金屬的電極電勢。
LiCoO2薄膜表面的電化學
作者研究了Li鹽/PC電解液中的鹽濃度和陰離子種類對LiCoO2薄膜電極電化學反應的影響。作者選擇LiCoO2薄膜作為電極,以避免添加劑(如聚合物粘合劑和碳導電劑)的影響。圖5a顯示了LiCoO2薄膜電極在[LiClO4]/[PC]=1/10電解液中的循環伏安圖。在大約3.9V處觀察到主要的氧化還原峰,這代表Li+在LiCoO2的層狀結構中的脫出或嵌入。
圖5.(a)[LiClO4]/[PC]=1/10和(b)[LiCl4]/[PC]=1/3電解液中LiCoO2薄膜電極下以1 mV s–1的掃描速率CV圖。
在摩爾比為[LiClO4]/[PC]=1/3的HCE中,還觀察到LiCoO2的可逆Li+嵌入/脫出反應(圖5b)。與[LiClO4]/[PC]=1/10電解液中相比,[LiClO3]/[PC]=1/3電解液中主要氧化還原峰的峰間距和伏安圖的峰電流密度分別較大和較低。這可能是由于前一種電解液的離子電導率(0.58 mS/cm)低于后一種電解液(5.59 mS/cm)。在[LiClO4]/[PC]=1/3電解液的情況下,抑制了循環時峰值電流密度的惡化,表明LiCoO2電極的穩定性隨著鹽濃度的增加而提高。稍后將進一步討論Li鹽濃度對LiCoO2電極穩定性的影響。
為了研究LiCoO2/電解液界面的電荷轉移反應動力學,進行了EIS測試。圖6顯示了不同電極電勢下[LiClO4]/[PC]=1/10電解液中LiCoO2薄膜電極的Nyquist 圖。在高頻范圍(>1kHz)中觀察到小半圓,其不隨電極電勢的變化而變化,這可能歸因于多晶LiCoO2膜中的電子電阻以及Au和LiCoO2之間的接觸電阻。
圖6.(a) LiCoO2薄膜電極(在[LiClO4]/[PC]=1/10電解液中在不同電極電位下的Nyquist 圖。(b)放大的Nyquist圖。
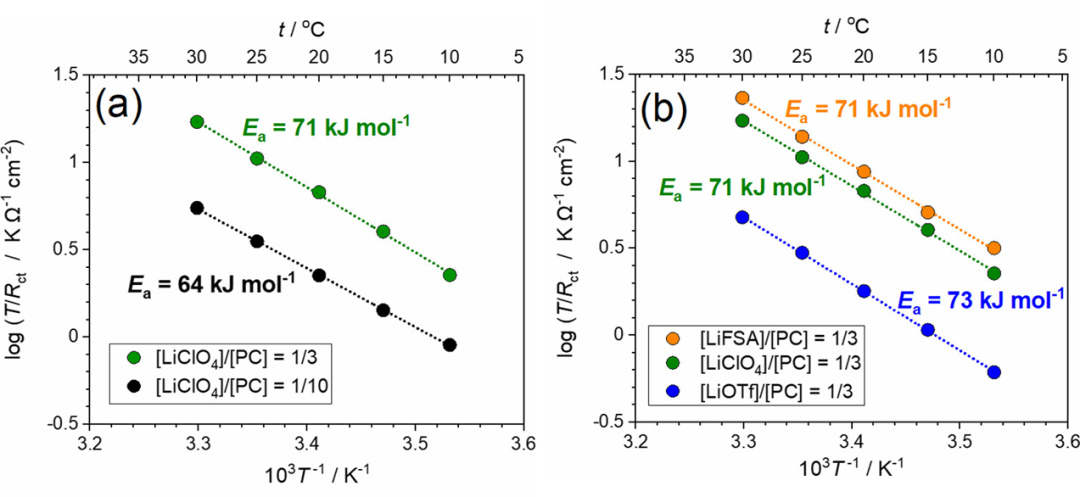
作者還研究了LiCoO2/電解液界面處Rct的溫度依賴性。圖7顯示了Li鹽/PC電解液中電極電位為4.0 V時T/Rct的Arrhenius圖。值得注意的是,在該溫度范圍內,[LiClO4]/[PC]=1/3電解液的Rct低于[LiClO4]/[PC]=1/10電解液的。Li+嵌入到部分脫鋰LizCoO2中的反應可以表示如下:
圖7. 在(a)[LiClO4]/[PC]=1/n(n=3,10)和(b)[LiX]/[PC]=1/3(X=FSA,ClO4,OTf)電解液中,LiCoO2薄膜電極在4.0 V下的T/Rct的Arrhenius圖。
Li+插層反應的交換電流密度i0與Rct成反比,可描述如下:
其中k0是標準速率常數,asolid是部分脫鋰LiCoO2中Li+的活度,α是轉移系數(0<α<1)。作者可以假設在4.0V的交流阻抗測量期間,部分脫鋰的LiCoO2中Li+的活度沒有變化。為了簡單起見,Li+的活度(aLi+)假設如下,即無論Li鹽濃度如何,自由溶劑的活度都不變(asolvent=1)(即,忽略電解液中溶劑活度的鹽濃度依賴性)。轉移系數α通常約為0.5。式4表明,i0隨著電解液中Li+離子活度(aLi+)(即Li鹽濃度)的增加而增加。標準速率常數k0表示如下:
其中A是指數前因子,ΔG*是活化的標準吉布斯能。文獻中報道,Li+嵌入反應的RDS是Li+在電極/電解液界面處的脫溶過程。因此,假設ΔG*主要來源于破壞Li+和溶劑之間的離子-偶極相互作用所需的活化勢壘。假設aLi+在測量的溫度范圍內不變,可以使用以下方程從Arrhenius圖(圖7a)估計Li+嵌入反應的表觀活化能Ea(app):
其中A′是指數前因子。然而,Ea(app)不等于ΔG*(見下文)。[LiClO4]/[PC]=1/10電解液的Ea(app)值估計為64 kJ mol–1,與之前報道的值相當。根據1/Rct的Arrhenius圖估計的表觀活化能為62 kJ/mol,這與圖7a所示的值沒有顯著差異。[LiClO4]/[PC]=1/3 HCE的Ea(app)值估計為71 kJ/mol,大于[LiClO4]/[PC]=1/10低濃度電解液(LCE)的Ea值,與之前報道的值相當。Abe報道了固體電解質/液態電解液和碳質電極/電解液界面的Ea(app)在3-5 M的高鹽濃度范圍內增加。他們推測,界面處Li+陰離子離子對的裂解需要比HCE中Li+離子脫溶更大的ΔG*。
除了活化吉布斯能ΔG*之外,電解液粘度(η)也對電極/電解液界面處的電荷轉移動力學有顯著影響。Uchimoto等人報道了LiMn2O4在恒溫下的Li+插層反應的Rct和η之間的線性關系。他們基于溶劑動力學理論解釋了這一點。如果溶劑動力學理論對Li+插層反應有效,則方程5中的指數前因子A將包括溶劑的縱向弛豫時間τL,并表示如下:
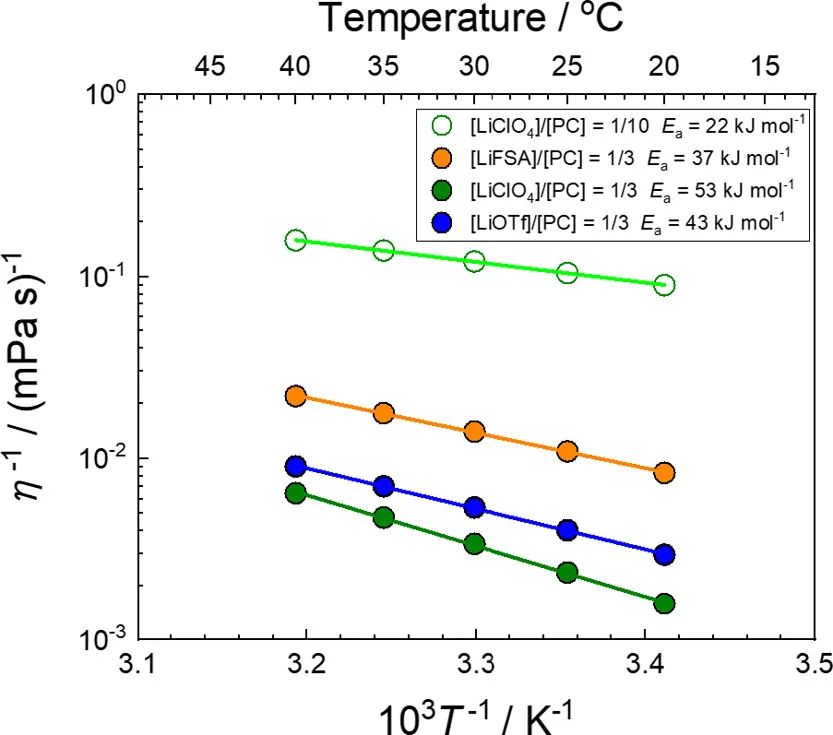
其中B=AτL。縱向弛豫時間τL與η大致成正比。(64)圖8顯示了基于PC的LCE和HCE的流動性(粘度的倒數,η–1)的阿倫尼烏斯圖。LCE具有比HCE更高的流動性(即更低的粘度)。在[Li鹽]/[PC]=1/3 HCE中, LiFSA的電解液表現出最高的流動性(圖8),這也可以解釋其高1/Rct(圖7b)。LiOTf基電解液的流動性高于LiClO4基電解液;然而,前者的1/Rct低于后者,可能是因為前者中Li+的活度系數較低(圖4)。隨著Li鹽濃度的增加,電解液溶液的流動性降低,而Li+活度增加。aLi+和η–1之間的折衷可以提供具有最大1/Rct值的電解液。
圖8. 組成為[LiFSA]/[PC]=1/3、[LiClO4]/[PC]=1/n(n=3,10)和[LiOTf]/[PC]=1/3的電解液的流動性(η–1)的阿倫尼烏斯圖。
盡管高濃電解液流動性的溫度依賴性在寬溫度范圍內遵循Vogel–Fulcher–Tammann方程,在20–40°C的窄溫度范圍內,流動性與1/T成線性比例。流動性的表觀活化能Ea(η–1)使用以下方程估算:
其中η0–1是指數前因子。如圖8所示,[Li鹽]/[PC]=1/3 HCE的Ea(η–1)值遠高于[LiClO4]/[PC]=1/10 LCE的值。考慮到τL的溫度依賴性,方程式7可改寫如下:
其中τ0–1是τL–1的Arrhenius方程中的指數前因子,Ea(τL–1)是τL–1的活化能。假設在測量溫度范圍內,τL與η成比例,則Ea(τL–1)等于Ea(η–1)。然而,隨著Li鹽濃度從[LiClO4]/[PC]=1/10增加到[LiClO4]/[PC]=1/3,Ea(app)的增加僅為7 kJ/mol(圖7a),而Ea(η–1)的增加為31 kJ/mol(圖8)。
總結與展望
作者研究了陰離子的Lewis堿性對LiCoO2薄膜電極在高濃PC電解液中的界面電荷轉移動力學的影響。隨著HCEs中陰離子的Lewis堿性降低,Li+與陰離子之間的離子締合減弱,游離PC的濃度降低,導致Li+的活度系數更高。EIS顯示,LiCoO2/電解液界面(Rct)處的電荷轉移電阻隨著電解液中Li+離子活度的增加而降低。
隨著Li鹽濃度的增加,游離PC的峰值強度降低,結合PC的峰值增加。對于[Li鹽]/[PC]=1/3的HCE和高度解離的鹽,如LiPF6和LiFSA,結合PC的峰值強度遠高于游離PC,這表明大多數PC分子與Li+離子配位,而游離PC分子幾乎不存在。相反,對于具有高度締合鹽(如LiBF4和LiOTf)的HCE,游離PC的峰值強度與結合PC的峰值相當或大于結合PC的強度,表明這些溶液中存在大量游離PC分子。
根據文獻,由于陰離子各自的Lewis堿性,離子締合的順序為LiPF6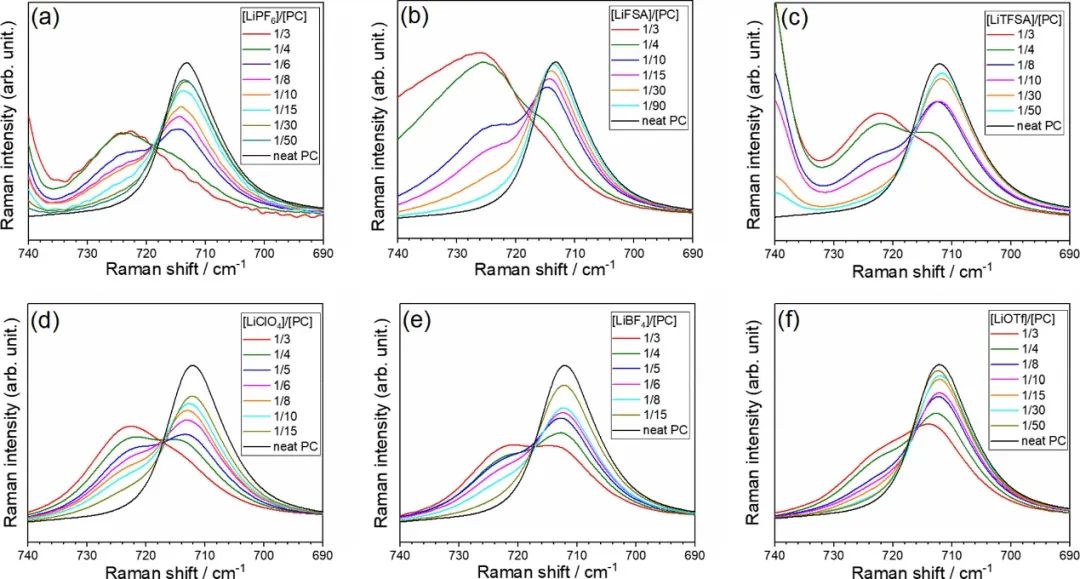
隨著LiClO4的摩爾分數增加到[LiClO4]/[PC]=1/3,在~940 cm–1處的肩峰變得更加突出,這表明ClO4–陰離子直接與Li+離子相互作用形成接觸離子對(CIP)。盡管在基于LiPF6、LiFSA和LiTFSA的電解液中,SSIP和CIP中涉及的陰離子的拉曼光譜帶重疊,但隨著Li鹽摩爾分數的增加,每個光譜帶向更高的波數移動,這表明Li+離子和陰離子之間的相互作用變得更強。
換句話說,無論陰離子種類如何,溶劑和陰離子都與HCE中的Li+離子競爭配位。由于Li+在非水電解液溶液中的配位數通常為4–5,電解液中的PC溶劑的量([Li鹽]/[PC]=1/3的摩爾比)不足以滿足該配位數。因此,Li+離子與陰離子配位并形成CIPs。
在[LiBF4]/[PC]=1/3和[LiOTf]/[PC]=1/3的混合物的拉曼光譜中,BF4-和OTf-的強峰分別出現在約775和約1050 cm-1處,表明形成了離子聚集體(AGG),其中陰離子與一個以上的Li+離子配位。這表明更多締合陰離子與多個Li+離子強烈相互作用,一定量的PC從Li+的第一溶劑化鞘中移除,導致電解液中游離PC的分數增加。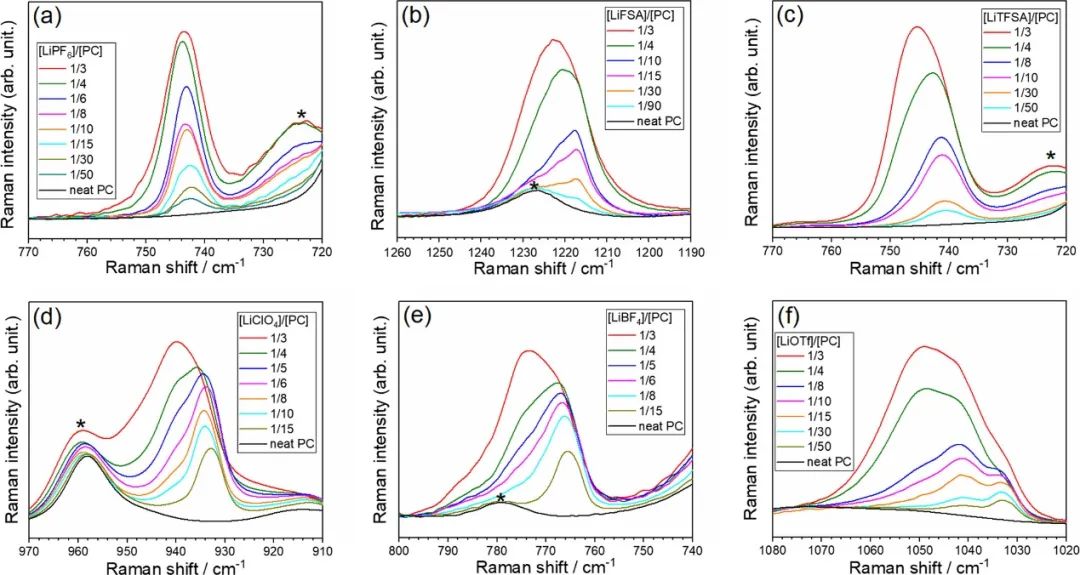
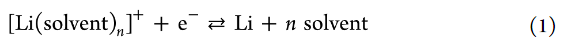

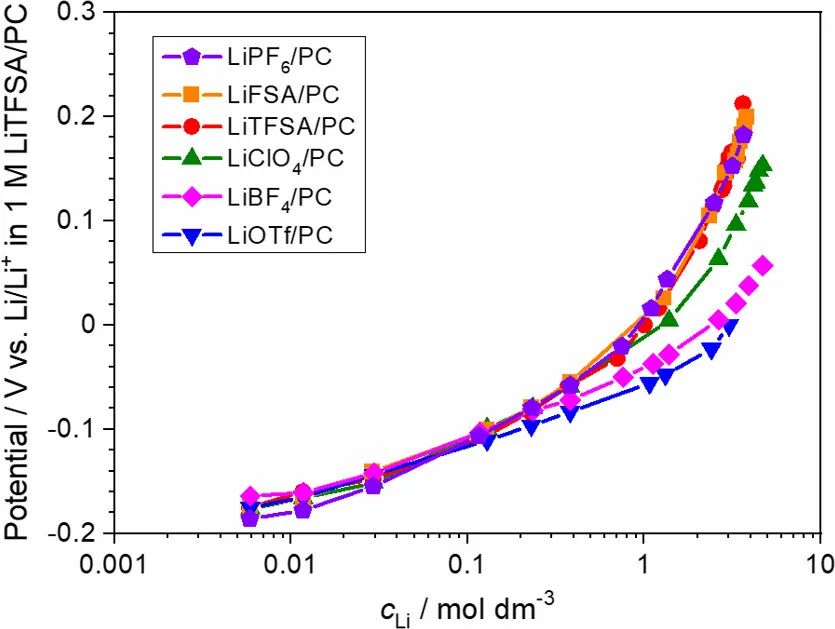
在含有過量溶劑的稀釋電解液溶液(≤0.1M)中,Li金屬的電極電勢隨log cLi線性增加,而與陰離子種類無關(圖4)。該結果表明,恒定溶劑活度的假設(即,a溶劑=1)對于稀電解液是有效的。在0.01–0.1 M的濃度范圍內,Li電極電勢與log cLi的斜率為50–60 mV/ decade,與陰離子種類無關,這與單電子反應的理論值(60 mV/decade)非常一致。
然而,對于高濃電解液(>1M和[Li鹽]/[PC]>1/8),Li電極電勢隨著鹽濃度的增加而非線性增加log cLi和Li電極電勢之間的非線性關系可歸因于Li+的活度系數(γ+)的增加(其中aLi+=γ+cLi)或自由溶劑活度的降低。實際上,如拉曼光譜所示,游離溶劑的濃度(或活度)隨著鹽濃度的增加而降低(圖2)。
然而,在傳統的電解液理論中,假設溶劑的活度不變(即,a溶劑=1),與鹽濃度無關。如果作者假設無論鹽濃度如何,溶劑的活度總是1(即忽略電解液中溶劑活度的鹽濃度依賴性),則在>1M Li鹽濃度下,Li+的活度系數應隨著cLi的增加而增加。
Li的電極電勢與log cLi的曲線斜率隨濃度范圍超過1M的陰離子而變化。這里,作者注意到陰離子配位能力的順序與離子締合強度有關,即PF6–
強配位陰離子優先與Li+相互作用,在高濃電解液中形成CIPs和AGG(圖3)。這可能導致具有強配位陰離子的高濃電解液中Li+的活度系數相對較低。即Li+的活度系數與陰離子的配位能力成反比。
在4.07和4.17V處出現兩個小峰,這歸因于CoO2框架中有序和無序Li+離子排列之間的相變。在1 mV s–1的掃描速率下,約3.9 V的主要氧化還原峰的峰相隔距離較小(約40 mV)然而,在循環時,峰值距離增加,峰值電流減少,表明在循環期間發生了LiCoO2多晶膜的開裂。在本研究中使用的電池配置的情況下,沒有向LiCoO2電極施加機械壓力。因此,如果在電化學反應期間膜發生破裂,則LiCoO2薄膜和Au襯底之間的電接觸可能部分惡化,導致一定數量的晶粒電失活。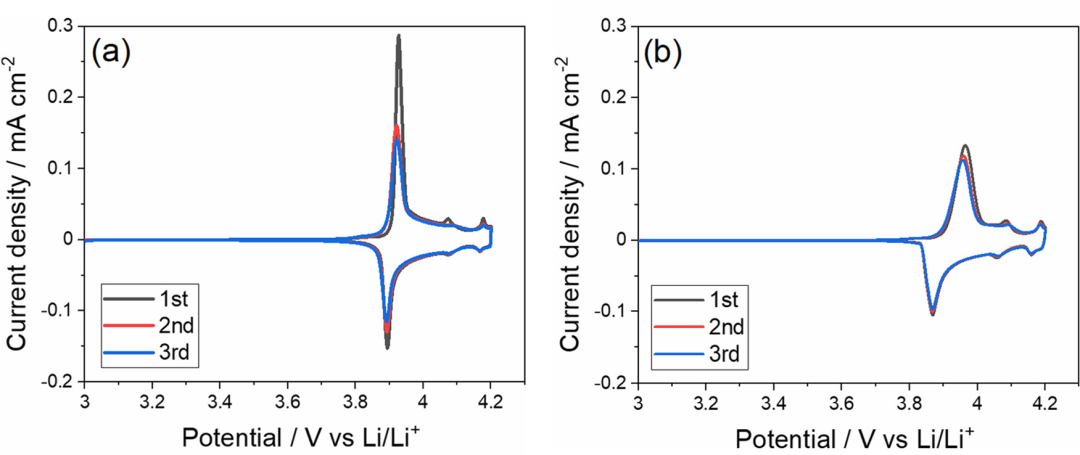
在3.4V的開路電位下,薄膜在<1kHz的頻率范圍內表現出電容行為(圖6a)。當電極電位高于3.8V時,在低頻區域中出現第二半圓形。該半圓的直徑隨著電極電勢的變化而變化,并在4.0V時達到最小值(圖6b)。因此,此半圓歸因于LiCoO2/電解液界面處的電荷轉移反應。然后,通過擬合圖6a中所示的等效電路來評估電荷轉移電阻(Rct),其中Rb是本體電解液的離子電阻,RHF是LiCoO2薄膜中的電子電阻,CPE是恒定相位元件,Zw是Warburg阻抗,CL是薄膜的Li+嵌入電容。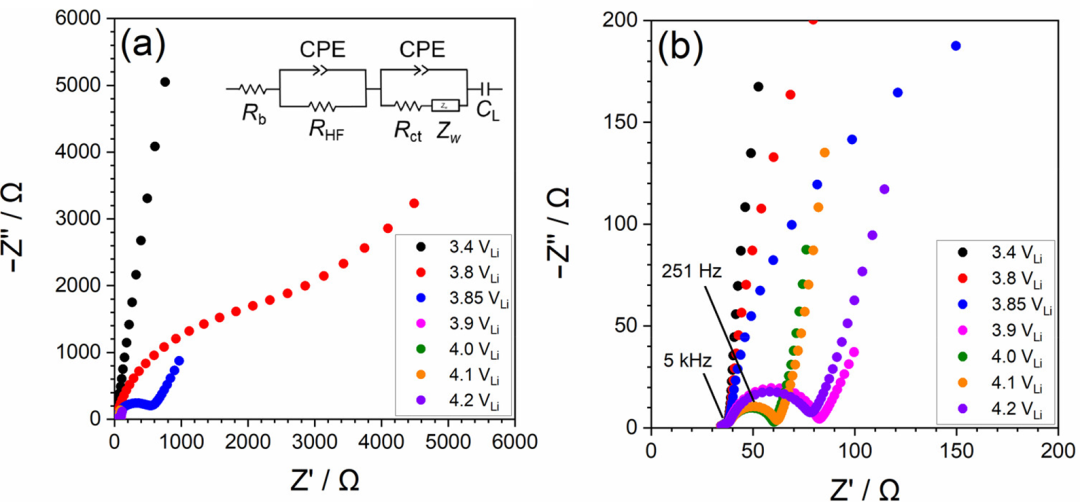
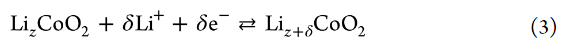
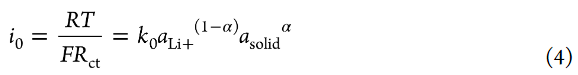

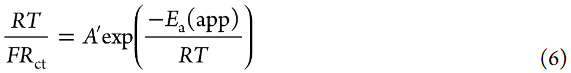
然而,這里出現了一個問題,即在所研究的電解液中,Li+和ClO4之間的吸引力是否比Li+和PC之間的吸引力強。古特曼供體數(DN)是Lewis堿性的描述符;PC的DN為15.1 kcal mol–1,而ClO4的DN為8.44 kcal/mol。由于PC是比ClO4更強的Lewis堿,Lewis酸性Li+離子主要被LCE中的PC分子溶劑化,如拉曼光譜所示(圖2)。在HCE中,如[LiClO4]/[PC]=1/3,PC和ClO4-都與Li+離子配位。如果Li+和陰離子之間的復合物形成是HCEs中Ea(app)增加的主要原因,則Ea(pp)預計會根據陰離子種類而變化。
為了證實這一假設,作者研究了含有不同Li鹽、LiFSA、LiClO4和LiOTf的[Li鹽]/[PC]=1/3電解液中Rct的溫度依賴性。如前所述,陰離子的配位能力按以下順序增加:FSA–
在30°C下測得的Rct值順序為FSA–
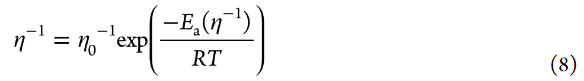
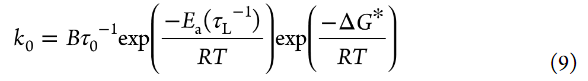
這種差異可能是由于其他因素的溫度依賴性。先前的文獻表明,電解液中Li+和陰離子之間的關聯隨著溫度的升高而增強。高溫下Li+和陰離子之間的增強關聯降低了Li+的活度系數(見上文),從而降低了Ea(app)值。此外,溶液中的陰離子種類影響Li+活度系數的溫度依賴性。因此,除了ΔG*,還有τL的溫度依賴性和Li+的活度系數影響HCE中的Ea(app)。
因此,在HCEs中使用具有弱Lewis堿性的陰離子的高度解離的Li鹽有利于在LiCoO2/電解液界面上實現快速的電荷轉移動力學。此外,游離PC的濃度影響電解液中部分脫鋰的LiCoO2電極的穩定性。在含有較高濃度游離PC的電解液中,部分脫鋰的LiCoO2電極的Rct和Li+嵌入電容(CL)分別隨時間增加和減少,而在含有較低濃度游離PC電解液中Rct增長和CL衰減減慢。因此,LiCoO2電極的穩定性可通過具有弱Lewis堿性的陰離子來改善。
審核編輯:劉清
-
鋰離子電池
+關注
關注
85文章
3254瀏覽量
77931 -
電解液
+關注
關注
10文章
852瀏覽量
23220 -
拉曼光譜
+關注
關注
0文章
86瀏覽量
2779 -
cip
+關注
關注
0文章
15瀏覽量
4753
原文標題:局部高濃先驅Kaoru Dokko!發文探討高濃電解液陰離子對Li+活度和嵌入反應動力學影響機理
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
強弱耦合型電解液調控超級電容器寬溫域特性及其機制研究

調控磷酸酯基阻燃電解液離子-偶極相互作用實現鈉離子軟包電池安全穩定運行
Li3MX6全固態鋰離子電池固體電解質材料
【Simcenter STAR-CCM+】通過快速準確的CFD仿真加速空氣動力學創新

水系電解液寬電壓窗口設計助力超長壽命水系鈉離子電池
北大潘鋒ACS Nano:高熵巖鹽表面層穩定超高鎳單晶正極
武漢理工大學在水系鋅離子電池研究方面取得新進展
鎳氫電池的電解液是什么
新宙邦擬在美國投建10萬噸/年電解液項目
單液原電池電流不穩定的原因是什么

非質子型弱配位電解液實現無腐蝕超薄鋅金屬電池






 工商網監
工商網監
評論